A Look Down Track B
I.
Depression probably has something to do with decreased synaptogenesis in the brain, maybe the hippocampus in particular. Neurons are less likely to respond to stimuli by connecting to other neurons. The whole network becomes sparser than usual, and dysfunctional thought-loops that thrive in sparse network conditions start taking over.
We understand parts of the pathways that regulate synaptic growth. When the body wants more synapses, it releases neurotrophic hormones like BDNF (brain-derived neurotrophic factor). These activate various receptors including tropomyosin receptor kinase B (TrkB, affectionately pronounced “track B” because it’s one of two related pathways for these signals). TrkB then something Ras mTORC something something synaptogenesis now you’re not depressed anymore hooray.
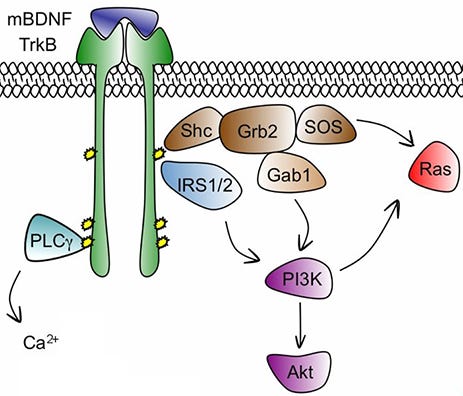 Pictured: BDNF binds to TrkB. The IRS confiscates 1/2 of it as taxes, which radicalizes the receptor and makes it join Gab (see footnote 1), where it tweets out an SOS message to the Ras of Ethiopia. But the left wing of the receptor joins the Palestine Liberation Council and moves to California (see footnotes 2+). California has sunshine and good beaches, so you stop feeling depressed.
Pictured: BDNF binds to TrkB. The IRS confiscates 1/2 of it as taxes, which radicalizes the receptor and makes it join Gab (see footnote 1), where it tweets out an SOS message to the Ras of Ethiopia. But the left wing of the receptor joins the Palestine Liberation Council and moves to California (see footnotes 2+). California has sunshine and good beaches, so you stop feeling depressed.
This part sort of makes sense. But it coexists uneasily with other puzzle pieces in our knowledge of depression. For example, we give people SSRIs, their serotonin levels go up, and this makes them feel better. Why? Because of BDNF something TrkB something mTORC something? Probably; mice with dysregulated BDNF/TrkB systems don’t benefit from antidepressants. But why does more serotonin cause BDNF something TrkB something? I’ve looked for years for a paper that says something like “by the way, serotonin makes cells release more BDNF”. But despite a few suggestive links I don’t see anyone strongly asserting that they understand this.
A recent study, Antidepressant drugs act by directly binding to TrkB neurotrophin receptors (h/t Derek Lowe) proposes a dramatic solution. The authors, mostly based out of a well-known lab in Helsinki, argue that antidepressants bind to TrkB directly, modifying it so that it responds more vigorously to BDNF. They explored the TrkB receptor in more detail than anyone had before, discovering some new functions (it binds to cholesterol) and a new binding site. They found that three antidepressants - fluoxetine (“Prozac”), imipramine, and ketamine, all bind to the new site (the textbooks say all three of these work in different ways). Three control non-antidepressant chemicals - diphenhydramine (“Benadryl”), chlorpromazine (“Thorazine”), and isoproterenol don’t. Once bound, the antidepressants seem to help TrkB receptors get to the parts of the cell where they can be useful. Then they show antidepressants don’t work on mice with a mutation that breaks TrkB’s ability to bind antidepressants (but not its ability to bind BDNF). In other words, the antidepressant effect was coming from direct antidepressant-TrkB binding, not serotonin -> BDNF -> TrkB binding! The serotonin doesn’t matter at all!
If this is true, how come antidepressants take about a month to work? This is a problem for the serotonin theory too - antidepressants start increasing serotonin immediately - but everyone has agreed that something something autoreceptor downregulation so probably it’s fine. What does the new theory say? It argues that it takes a lot of antidepressant to activate TrkB - so much that it takes a month for the right amount to accumulate in the brain (wait, antidepressants accumulate in the brain over months? we’ll come back to this part.)
II.
I’m not a cellular biologist or a biochemist, so I can’t comment on most of the high-tech methods used in this paper. All I can do is give a psychiatrist’s response, informed by working with some of these substances in real life. This is a great theory, one that would be the biggest psychiatric breakthrough in a decade if true. But I’m not buying it yet.
First off: if antidepressants’ effects are unrelated to monoamines, why are so many antidepressants monoaminergic? Actually, that’s the wrong question, if pharma companies only looked for monoamine antidepressants, that’s all they’d find. But why are so many monoaminergic substances antidepressants? Where are all the papers saying “we tested this new SSRI, it did a great job inhibiting serotonin reuptake, but there was no antidepressant effect whatsoever”? Surely some substances that are SSRIs don’t also interact with this new TrkB receptor domain? Are pharma companies hiding their failures? But if they kept failing, surely they themselves would realize something was up and stop throwing money at serotonergic antidepressants? So many people for so many years have been trying to poke holes in the serotonergic model of antidepressants, and they’ve made some good points, but I’ve never seen any of them say “and here are these fifty chemicals which are great SSRIs but totally useless for depression”. Why not?
And how come 5-HTP and l-tryptophan, two supplements which are just serotonin precursors, seem to help depression? How come mesembrine seems to help depression, and we didn’t know why, and then we checked and found it inhibited SERT? How come depleting animals (or people) of serotonin precursors makes them depressed? How come every strong MAOI is a great antidepressant, their antidepressant strength is proportional to their MAOI strength, and when we take random MAOIs and check if they work for depression, they do? Do all these monoaminergic substances, by coincidence, also bind to TrkB?
The TrkB paper cites two papers saying antidepressants still work on mice genetically engineered so they can’t reuptake serotonin, which would mean serotonin isn’t involved in their effects and it’s probably this new TrkB thing. They also cite one paper saying the antidepressants don’t work on the mice, just as the standard theory would predict. They imply this means it’s two to one, but reviews of this literature make it sound like the antidepressants not working is the more common finding, and when I Google around on this subject most papers seem to agree.
 Alex Nackenoff, Ph.D. @anackenoff@zenbrainest @Dereklowe (excuse my self-promotion) We did show that SERT M172 mice lost acute and chronic SSRI behavioral and neurogenic efficacy, which should retain all TrkB effects. Thus TrkB effects are insufficient alone to produce AD effects.
Alex Nackenoff, Ph.D. @anackenoff@zenbrainest @Dereklowe (excuse my self-promotion) We did show that SERT M172 mice lost acute and chronic SSRI behavioral and neurogenic efficacy, which should retain all TrkB effects. Thus TrkB effects are insufficient alone to produce AD effects.  pubmed.ncbi.nlm.nih.govEssential Contributions of Serotonin Transporter Inhibition to the Acute and Chronic Actions of Fluoxetine and Citalopram in the SERT Met17…Depression is a common mental illness and a leading cause of disability. The most widely prescribed antidepressant medications are serotonin (5-HT) selective reuptake inhibitors (SSRIs). Although there is much support for 5-HT transporter (SERT) antagonism as a basis of antidepressant efficacy, this…[5:18 PM ∙ Feb 19, 2021
pubmed.ncbi.nlm.nih.govEssential Contributions of Serotonin Transporter Inhibition to the Acute and Chronic Actions of Fluoxetine and Citalopram in the SERT Met17…Depression is a common mental illness and a leading cause of disability. The most widely prescribed antidepressant medications are serotonin (5-HT) selective reuptake inhibitors (SSRIs). Although there is much support for 5-HT transporter (SERT) antagonism as a basis of antidepressant efficacy, this…[5:18 PM ∙ Feb 19, 2021
15Likes3Retweets](https://twitter.com/anackenoff/status/1362813927709679624)
Likewise, there are a lot of papers showing that antidepressant treatment increases levels of BDNF and that this seems to be necessary for its effects. If antidepressants just positive-allosteric-modulated TrkB, we’d expect to see the same amount of (maybe less) BDNF.
There are maybe a few hundred things like this? I promise you that psychiatrists haven’t forgotten to check whether serotonin is involved in the mechanism of SSRIs? It’s been a pretty big deal for us?
This doesn’t mean the psychiatric community can’t be wrong. It could be the same sort of thing as 5-HTTLPR, another subject where there were dozens of studies establishing an effect, replicating it, and explaining all the little details of how it worked, before we realized they were all wrong and the effect didn’t exist. In fact, a version of this essay ten years ago would have listed 5-HTTLPR polymorphisms affecting antidepressant response as some of the evidence that antidepressant response was serotonergic! It could absolutely be true that all of these hundreds of studies were wrong! But it’s also possible that this one new study nobody has had a chance to check very well yet is wrong, and usually that’s your default assumption.
(The study does admit “it is unlikely that effects on TRKB mediate all the effects of ADs” and that “inhibition of 5HTT [serotonin transporters] and NMDA receptors also play a role”, but that doesn’t really seem to fit some of their other claims - like with the knockout mice - and feels kind of like hedging.)
(Also, all these mice studies are great, but in human studies, SSRIs barely work. The best meta-analyses of thousands of depressed patients manage to pick up a small signal, assuming the pharma companies haven’t figured out some way to fake us all out. But all these animal researchers are like “We genetically engineered a dozen mice to lack some gene, and tested whether SSRIs worked on them by seeing how sad their little mouse faces looked, and they worked great!” If you can detect SSRIs working on your dozen mice, how come we can only barely detect them in our several thousand humans who fill out long complicated depression tests? I think the answer is something like all the mice are genetically near-identical and kept in near-identical situations, and sometimes you literally breed mice for being SSRI responders. But I am still suspicious.)
Anyway, I promised I’d get back to the idea of antidepressants eventually accumulating over months until they reach the level where they can affect TrkB. I have been reading stuff about antidepressants for years and never heard anything about them accumulating like this? I mean, they accumulate over five or six days as you wait for them to reach plasma steady state, but that’s very different from the month or two it takes to work. Fluoxetine does accumulate for a month or two, just because of its half-life, but since other SSRIs aren’t like that, it’s probably not responsible for class-wide features.
 Source here
Source here
The TrkB paper links to two studies to make its claim that antidepressants typically accumulate in the brain over long periods. One is about fluoxetine; everyone knows fluoxetine does this, but it’s the only one and shouldn’t be considered typical. The other is a theoretical 1995 paper saying that a non-antidepressant, amantadine, accumulates in cellular lysosomes in a way that has unclear clinical implications, and maybe antidepressants could also do this? As far as I know, nobody has taken these people up on this suggestion in the past 25 years, and every subsequent study has shown antidepressants reach the brain in a pretty normal way. The authors of the TrkB study are either so far beyond me in their knowledge of obscure lysosomal accumulation methods that I am totally unqualified to assess their work, or else making a kind of rookie mistake where they think everything accumulates as slowly as fluoxetine does. Based on them kind of eliding the difference between the fluoxetine studies and the lysosome speculation, I think it’s the rookie mistake (though I’m not sure, and I promise to be suitably embarrassed if they blow my mind with interesting lysosome facts).
Okay, that’s me wading into a field I know nothing about and criticizing my betters. What do the actual biochemists and cell biologists say? There’s been some good discussion on Derek Lowe’s Twitter:
 Keri Martinowich @martinowk
Keri Martinowich @martinowk@zenbrainest @Dereklowe I’m not commenting on the Cell paper. But, just noting, as someone very familiar with neurotrophin research, that our field has been fraught w/ issues trying to assess TrkB binding and agonism See: stke.sciencemag.org/content/10/493… and:
 blogs.sciencemag.orgThose Compounds Aren’t What You Think They AreThere’s a lot of work in the literature on the TrkB receptor, which responds to brain-derived neutrotrophic factor (BDNF). That name certainly makes the ligand protein sound like a pretty big deal, and so it is: BDNF is involved in a lot of neural development pathways, injuries to nerve tissue, and
blogs.sciencemag.orgThose Compounds Aren’t What You Think They AreThere’s a lot of work in the literature on the TrkB receptor, which responds to brain-derived neutrotrophic factor (BDNF). That name certainly makes the ligand protein sound like a pretty big deal, and so it is: BDNF is involved in a lot of neural development pathways, injuries to nerve tissue, and{kind=link}
32Likes2Retweets](https://twitter.com/martinowk/status/1362823295502983181)
 Weinshenker Lab @LCneuroscience@samuelkohtala @zenbrainest @eperlste @Dereklowe TrkB assays probably very finicky and sensitive to small variations in protocol, compound batch, etc. Would be helpful to have coordinated replication efforts by multiple labs using same cells, antibodies, compound batch, etc. Crystal structure/cryo-EM of proteins + drug too.4:44 PM ∙ Feb 20, 2021
Weinshenker Lab @LCneuroscience@samuelkohtala @zenbrainest @eperlste @Dereklowe TrkB assays probably very finicky and sensitive to small variations in protocol, compound batch, etc. Would be helpful to have coordinated replication efforts by multiple labs using same cells, antibodies, compound batch, etc. Crystal structure/cryo-EM of proteins + drug too.4:44 PM ∙ Feb 20, 2021
 Samuel Kohtala @samuelkohtala@eperlste @LCneuroscience @zenbrainest @Dereklowe I don’t think anyone here is trying to toss any result out. But having worked with TrkB signaling for the past 6 years or so, I know there are a lot of issues in the field. Biological and non-biological.5:05 PM ∙ Feb 20, 2021
Samuel Kohtala @samuelkohtala@eperlste @LCneuroscience @zenbrainest @Dereklowe I don’t think anyone here is trying to toss any result out. But having worked with TrkB signaling for the past 6 years or so, I know there are a lot of issues in the field. Biological and non-biological.5:05 PM ∙ Feb 20, 2021
They say TrkB is really hard to work with and tends to produce lots of false positives - see especially this study.
Based on all this, I’m going to stick with the usual SSRIs -> serotonin -> BDNF -> TrkB -> nerve growth theory for now, but I look forward to seeing further attempts to replicate and explicate these interesting results.
**Prediction:** 90% chance that no major academic or popular resource (eg UpToDate, NICE, Wikipedia, WebMD) will give this theory prominence equal to or greater than the standard serotonin theory in its explanation of antidepressant effects by 2030, unless this is because some third theory has obsoleted both of them and neither gets any space.
**Prediction:** 95% chance that no drug with a design directly based on this theory will be approved for depression by the FDA by 2030